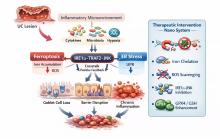
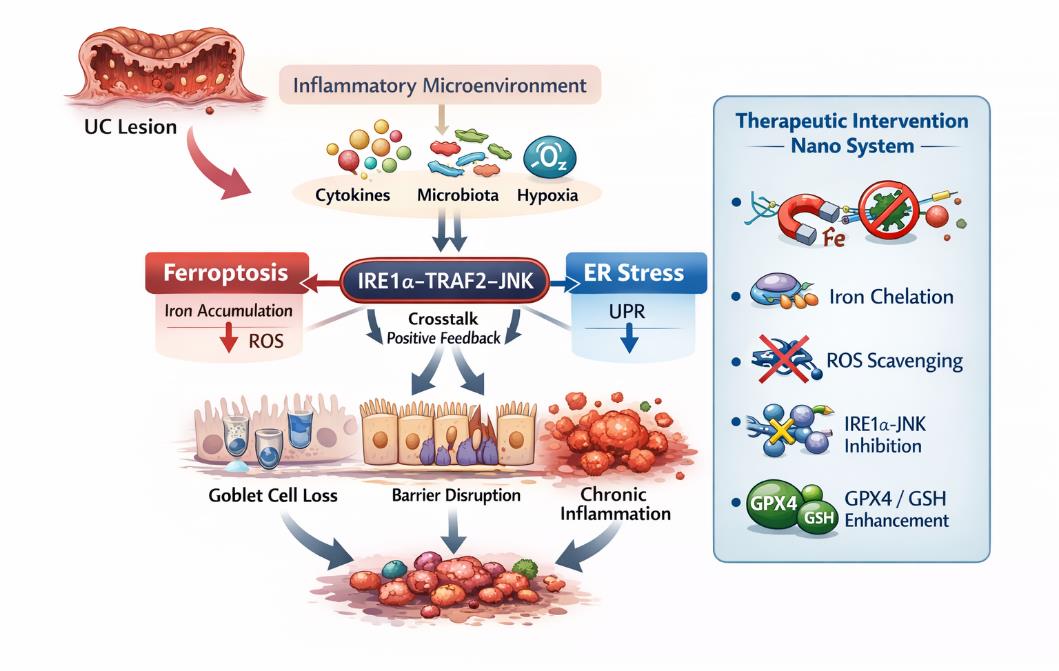
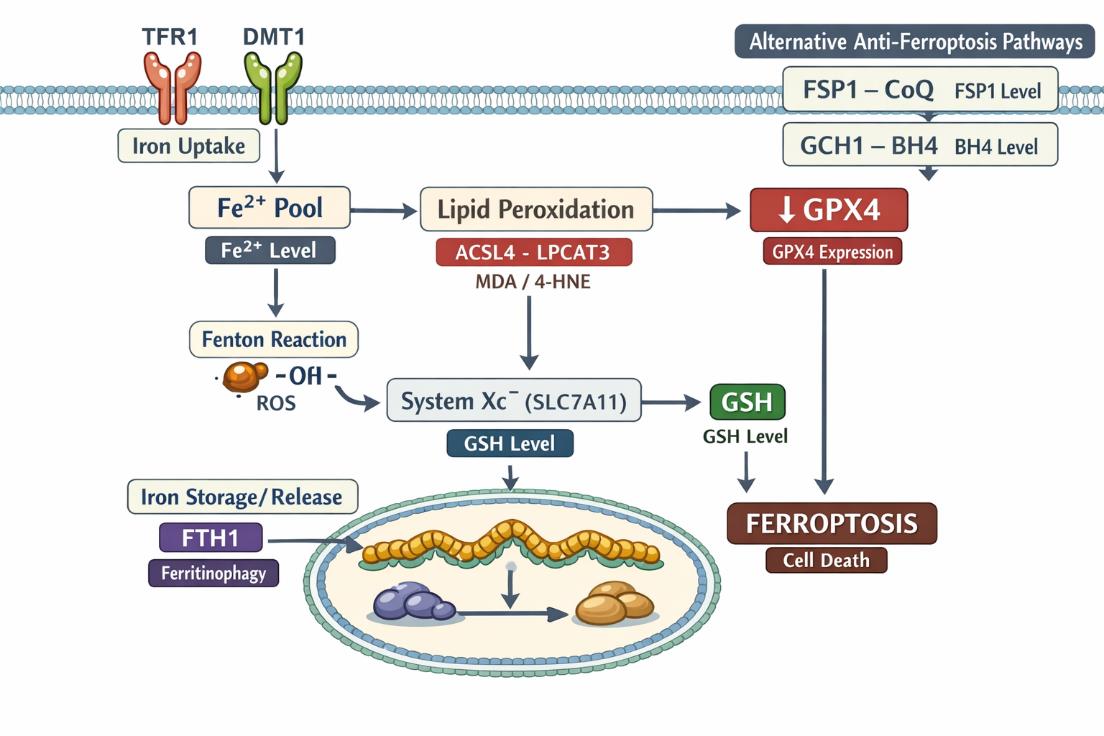
| [1] |
XU J, LIU S, CUI Z, et al. Ferrostatin-1 alleviated TNBS induced colitis via the inhibition of ferroptosis[J]. Biochem Biophys Res Commun, 2021, 573: 48-54. doi:10.1016/j.bbrc. 2021.08.018 .
doi: 10.1016/j.bbrc. 2021.08.018
|
| [2] |
KASER A, LEE A H, FRANKE A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease[J]. Cell, 2008, 134(5): 743-756. doi:10.1016/j.cell.2008.07.021 .
doi: 10.1016/j.cell.2008.07.021
|
| [3] |
ZHANG H-S, CHEN Y, FAN L, et al. The endoplasmic reticulum stress sensor IRE1α in intestinal epithelial cells is essential for protecting against colitis[J]. J Biol Chem, 2015, 290(24): 15327-15336. doi:10.1074/jbc.M114.633560 .
doi: 10.1074/jbc.M114.633560
|
| [4] |
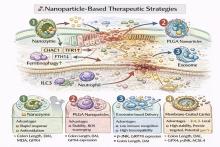
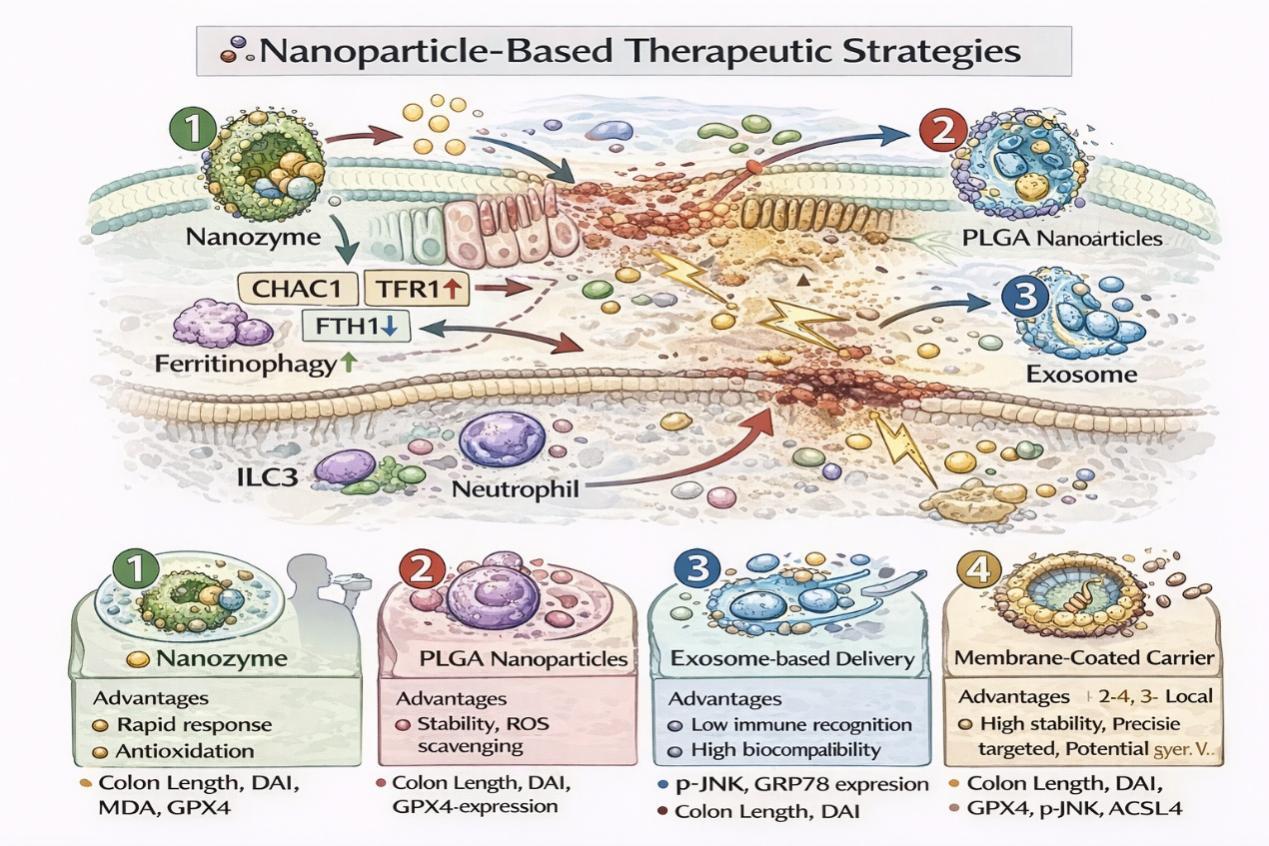
DONG H, DU W, DONG J, et al. Depletable peroxidase-like activity of Fe3O4 nanozymes accompanied with separate migration of electrons and iron ions[J]. Nat Commun, 2022, 13(1):5365. doi:10.1038/s41467-022-33098-y .
doi: 10.1038/s41467-022-33098-y
|
| [5] |
HONG Y, BOITI A, VALLONE D, et al. Reactive oxygen species signaling and oxidative stress: Transcriptional regulation and evolution[J]. Antioxidants, 2024, 13(3): 312. doi:10.3390/antiox13030312 .
doi: 10.3390/antiox13030312
|
| [6] |
CUI X, ZHANG Y, LU Y, et al. ROS and endoplasmic reticulum stress in pulmonary disease[J]. Front Pharmacol, 2022, 13: 879204. doi:10.3389/fphar.2022.879204 .
doi: 10.3389/fphar.2022.879204
|
| [7] |
DIXON S J, LEMBERG K M, LAMPRECHT M R, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death[J]. Cell, 2012, 149(5): 1060-1072. doi:10.1016/j.cell. 2012. 03.042 .
doi: 10.1016/j.cell. 2012. 03.042
|
| [8] |
STOCKWELL B R, JIANG X, GU W, et al. Emerging mechanisms and disease relevance of ferroptosis[J]. Trends Cell Biol, 2020, 30(6): 478-490. doi:10.1016/j.tcb.2020.02.009 .
doi: 10.1016/j.tcb.2020.02.009
|
| [9] |
FRIEDMANN ANGELI J P, SCHNEIDER M, PRONETH B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice[J]. Nat Cell Biol, 2014, 16(12): 1180-1191. doi:10.1038/ncb3064 .
doi: 10.1038/ncb3064
|
| [10] |
MURPHY M E. Ironing out how P53 regulates ferroptosis[J]. Proc Natl Acad Sci U S A, 2016, 113(44): 12350-12352. doi:10.1073/pnas.1615159113 .
doi: 10.1073/pnas.1615159113
|
| [11] |
HUANG J, ZHANG J, MA J, et al. Inhibiting ferroptosis: A novel approach for ulcerative colitis therapeutics[J]. Oxid Med Cell Longev, 2022, 2022:9678625. doi:10.1155/2022/9678625 .
doi: 10.1155/2022/9678625
|
| [12] |
XIE H, CAO C, SHU D, et al. The important role of ferroptosis in inflammatory bowel disease[J]. Front Med, 2024, 11: 1449037. doi:10.3389/fmed.2024.1449037 .
doi: 10.3389/fmed.2024.1449037
|
| [13] |
XU M, TAO J, YANG Y, et al. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis[J]. Cell Death Dis, 2020, 11(2):86. doi:10.1038/s41419-020-2299-1 .
doi: 10.1038/s41419-020-2299-1
|
| [14] |
LI X, HE J, GAO X, et al. GPX4 restricts ferroptosis of NKp46+ ILC3s to control intestinal inflammation[J]. Cell Death Dis, 2024, 15(9):687. doi:10.1038/s41419-024-07060-3 .
doi: 10.1038/s41419-024-07060-3
|
| [15] |
CHEN H, QIAN Y, JIANG C, et al. Butyrate ameliorated ferroptosis in ulcerative colitis through modulating Nrf2/GPX4 signal pathway and improving intestinal barrier[J]. Biochim Biophys Acta Mol Basis Dis, 2024, 1870(2): 166984. doi:10.1016/j.bbadis.2023.166984 .
doi: 10.1016/j.bbadis.2023.166984
|
| [16] |
ZHENG S, YIN J, WANG B, et al. Polydatin protects against DSS-induced ulcerative colitis via Nrf2/Slc7a11/Gpx4-dependent inhibition of ferroptosis signalling activation[J]. Front Pharmacol, 2025, 15:1513020. doi:10.3389/fphar.2024.1513020 .
doi: 10.3389/fphar.2024.1513020
|
| [17] |
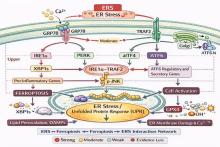
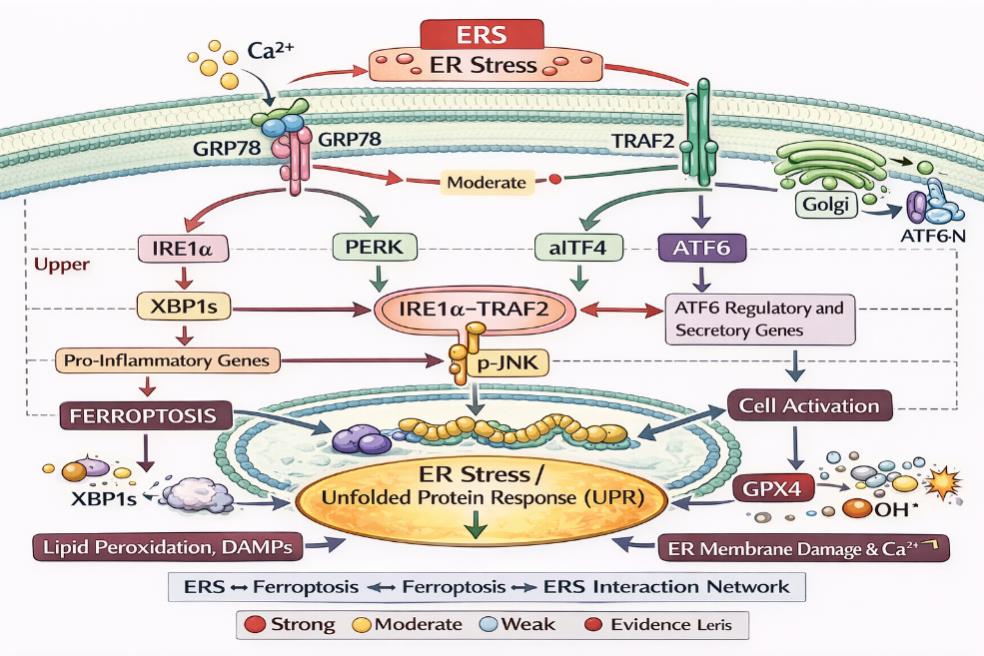
HETZ C, PAPA F R. The unfolded protein response and cell fate control[J]. Mol Cell, 2018, 69(2): 169-181. doi:10.1016/j.molcel.2017.06.017 .
doi: 10.1016/j.molcel.2017.06.017
|
| [18] |
COLEMAN O I, HALLER D, et al. ER stress and the UPR in shaping intestinal tissue homeostasis and immunity[J]. Front Immunol, 2019, 10:2825. doi:10.3389/fimmu.2019.02825 .
doi: 10.3389/fimmu.2019.02825
|
| [19] |
RODRIGUES B L, DOTTI I, PASCOAL L B, et al. Endoplasmic reticulum stress in colonic mucosa of ulcerative colitis patients is mediated by PERK and IRE1 pathway activation[J]. Mediators Inflamm, 2022, 2022: 1-13. doi:10.1155/2022/6049500 .
doi: 10.1155/2022/6049500
|
| [20] |
TRÉTON X, PÉDRUZZI E, CAZALS-HATEM D, et al. Altered endoplasmic reticulum stress affects translation in inactive colon tissue from patients with ulcerative colitis[J]. Gastroenterology, 2011, 141(3): 1024-1035. doi:10.1053/j.gastro.2011.05.033 .
doi: 10.1053/j.gastro.2011.05.033
|
| [21] |
JEONG H, HONG E H, AHN J H, et al. ERdj5 protects goblet cells from endoplasmic reticulum stress-mediated apoptosis under inflammatory conditions[J]. Exp Mol Med, 2023, 55(2): 401-412. doi:10.1038/s12276-023-00945-x .
doi: 10.1038/s12276-023-00945-x
|
| [22] |
WANG Y, HAO Y, YUAN L, et al. Ferroptosis: A new mechanism of traditional Chinese medicine for treating ulcerative colitis[J]. Front Pharmacol, 2024, 15:1379058. doi:10.3389/fphar. 2024.1379058 .
doi: 10.3389/fphar. 2024.1379058
|
| [23] |
OCANSEY D, YUAN J, WEI Z, et al. Role of ferroptosis in the pathogenesis and as a therapeutic target of inflammatory bowel disease (review)[J]. Int J Mol Med, 2023, 51(6):53. doi:10.3892/ijmm.2023.5256 .
doi: 10.3892/ijmm.2023.5256
|
| [24] |
FAN X, LU Q, JIA Q, et al. Prevotella histicola ameliorates DSS-induced colitis by inhibiting IRE1α-JNK pathway of ER stress and NF-κB signaling[J]. Int Immunopharmacol, 2024, 135: 112285. doi:10.1016/j.intimp.2024.112285 .
doi: 10.1016/j.intimp.2024.112285
|
| [25] |
URANO F, WANG X, BERTOLOTTI A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1[J]. Science, 2000, 287(5453): 664-666. doi:10.1126/science.287.5453.664 .
doi: 10.1126/science.287.5453.664
|
| [26] |
YANG J, WANG Y, LIU F, et al. Crosstalk between ferroptosis and endoplasmic reticulum stress: A potential target for ovarian cancer therapy (review)[J]. Int J Mol Med, 2025, 55(6): 1-20. doi:10.3892/ijmm.2025.5538 .
doi: 10.3892/ijmm.2025.5538
|
| [27] |
HUO C, LI G, HU Y, et al. The impacts of iron overload and ferroptosis on intestinal mucosal homeostasis and inflammation[J]. Int J Mol Sci, 2022, 23(22): 14195. doi:10.3390/ijms 232214195 .
doi: 10.3390/ijms 232214195
|
| [28] |
YAO T, DONG X, LV J, et al. Propionate alleviated colitis by modulating iron homeostasis to inhibit ferroptosis and macrophage polarization[J]. Int Immunopharmacol, 2025, 162: 115151. doi:10.1016/j.intimp.2025.115151 .
doi: 10.1016/j.intimp.2025.115151
|
| [29] |
MINOR E A, KUPEC J T, NICKERSON A J, et al. Increased DMT1 and FPN1 expression with enhanced iron absorption in ulcerative colitis human colon[J]. Am J Physiol Cell Physiol, 2020, 318(2): C263-C271. doi:10.1152/ajpcell.00128.2019 .
doi: 10.1152/ajpcell.00128.2019
|
| [30] |
LIU Z, ARCOS M, MARTIN D R, et al. Myeloid FTH1 deficiency protects mice from colitis and colitis-associated colorectal cancer via reducing DMT1-imported iron and STAT3 activation[J]. Inflamm Bowel Dis, 2023, 29(8): 1285-1296. doi:10.1093/ibd/izad009 .
doi: 10.1093/ibd/izad009
|
| [31] |
CHEN Y, ZHANG P, CHEN W, et al. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway[J]. Immunol Lett, 2020, 225: 9-15. doi:10.1016/j.imlet.2020.06.005 .
doi: 10.1016/j.imlet.2020.06.005
|
| [32] |
GUO Z, CHI R, PENG Y, et al. The role and interactive mechanism of endoplasmic reticulum stress and ferroptosis in musculoskeletal disorders[J]. Biomolecules, 2024, 14(11): 1369. doi:10.3390/biom14111369 .
doi: 10.3390/biom14111369
|
| [33] |
GU K, WU A, YU B, et al. Iron overload induces colitis by modulating ferroptosis and interfering gut microbiota in mice[J]. Sci Total Environ, 2023, 905: 167043. doi:10.1016/j.scitotenv.2023.167043 .
doi: 10.1016/j.scitotenv.2023.167043
|
| [34] |
SUN J, WANG Y, DU Y, et al. Involvement of the JNK/HO-1/FTH1 signaling pathway in nanoplastic-induced inflammation and ferroptosis of BV2 microglia cells[J]. Int J Mol Med, 2023, 52(1). doi:10.3892/ijmm.2023.5264 .
doi: 10.3892/ijmm.2023.5264
|
| [35] |
CHEN H, SUN W, LI C, et al. Inflammatory targeted nanoplatform incorporated with antioxidative nano iron oxide to attenuate ulcerative colitis progression[J]. iScience, 2025, 28(5): 112448. doi:10.1016/j.isci.2025.112448 .
doi: 10.1016/j.isci.2025.112448
|
| [36] |
FENG Y, LUO X, LI Z, et al. A ferroptosis-targeting ceria anchored halloysite as orally drug delivery system for radiation colitis therapy[J]. Nat Commun, 2023, 14(1):5083. doi:10.1038/s41467-023-40794-w .
doi: 10.1038/s41467-023-40794-w
|
| [37] |
WANG H, LIU Y, HUANG R, et al. Oral HMPB/Mn3O4 nanozymes for the treatment of ulcerative colitis via cellular redox balance regulation, gut microbiota modulation, and ferroptosis inhibition[J]. Chem Eng J, 2025, 519: 165265. doi:10.1016/j.cej.2025.165265 .
doi: 10.1016/j.cej.2025.165265
|
| [38] |
FAN S, ZHAO Y, YAO Y, et al. Oral colon-targeted pH-responsive polymeric nanoparticles loading naringin for enhanced ulcerative colitis therapy[J]. J Transl Med, 2024, 22(1):878. doi:10.1186/s12967-024-05662-1 .
doi: 10.1186/s12967-024-05662-1
|
| [39] |
SHI J, ZHOU J, LIU B, et al. Enzyme/ROS dual-sensitive nanoplatform with on-demand celastrol release capacity for enhanced ulcerative colitis therapy by ROS scavenging, microbiota rebalancing, inflammation alleviating[J]. J Nanobiotechnol, 2024, 22(1):437. doi:10.1186/s12951-024-02725-9 .
doi: 10.1186/s12951-024-02725-9
|
| [40] |
ZHOU J, LU P, HE H, et al. The metabolites of gut microbiota: their role in ferroptosis in inflammatory bowel disease[J]. Eur J Med Res, 2025, 30(1):248. doi:10.1186/s40001-025-02524-4 .
doi: 10.1186/s40001-025-02524-4
|


 )
)