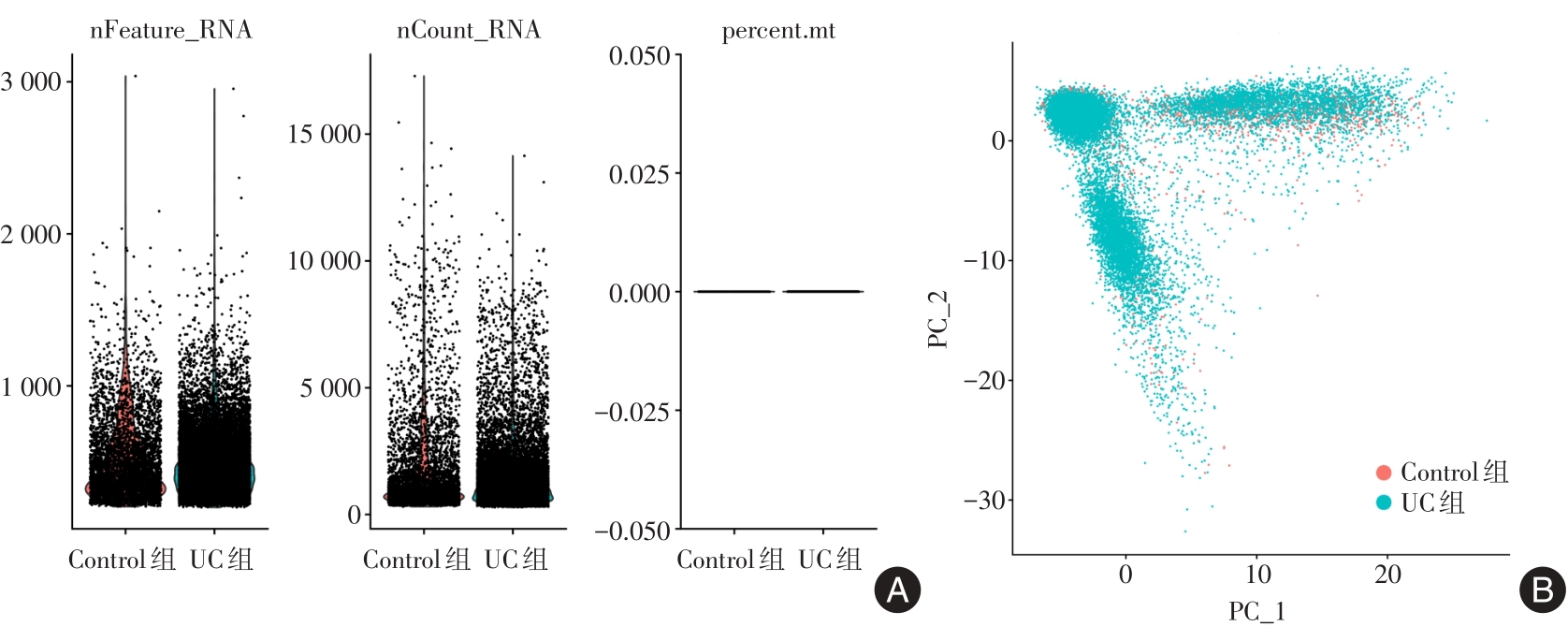
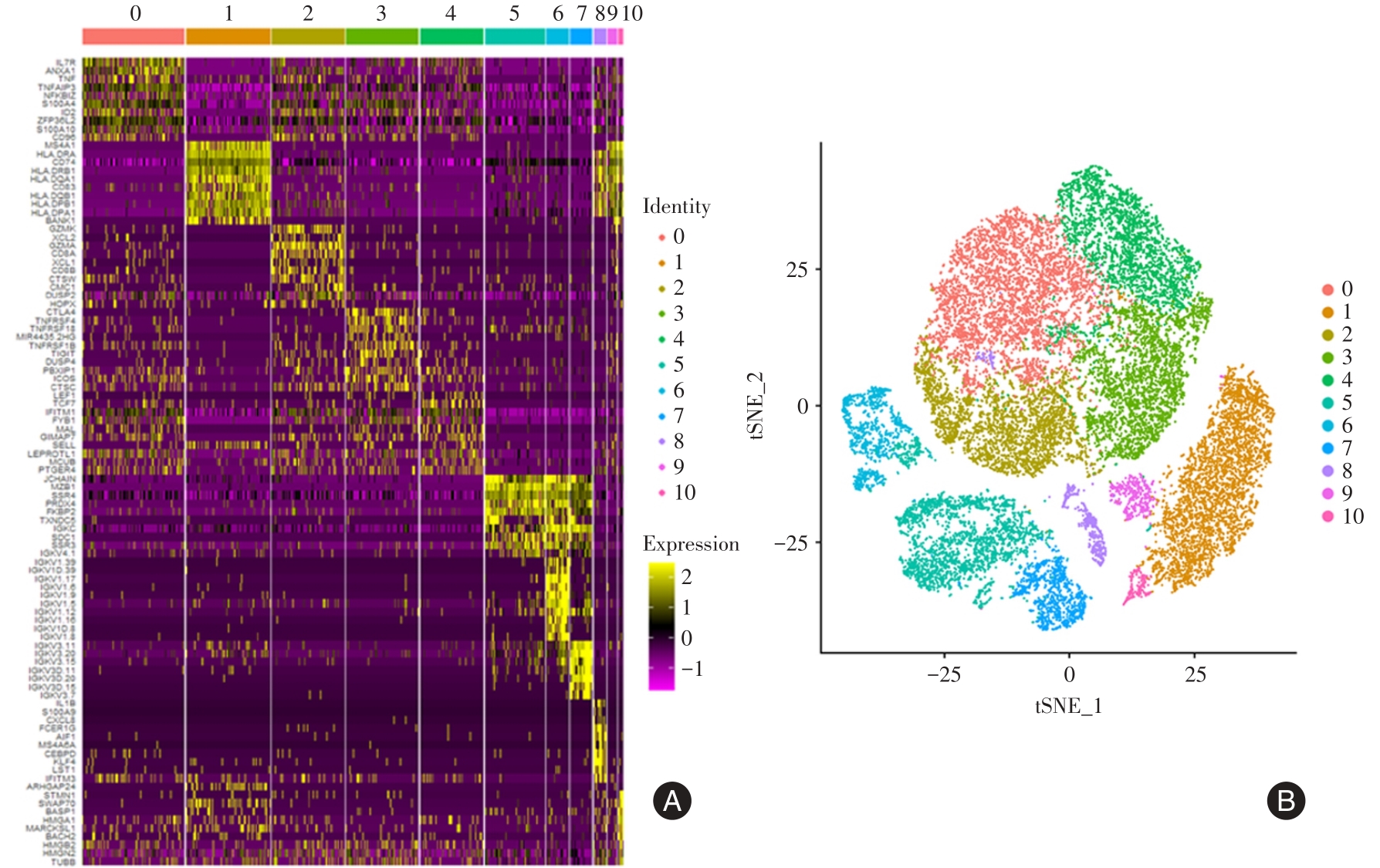
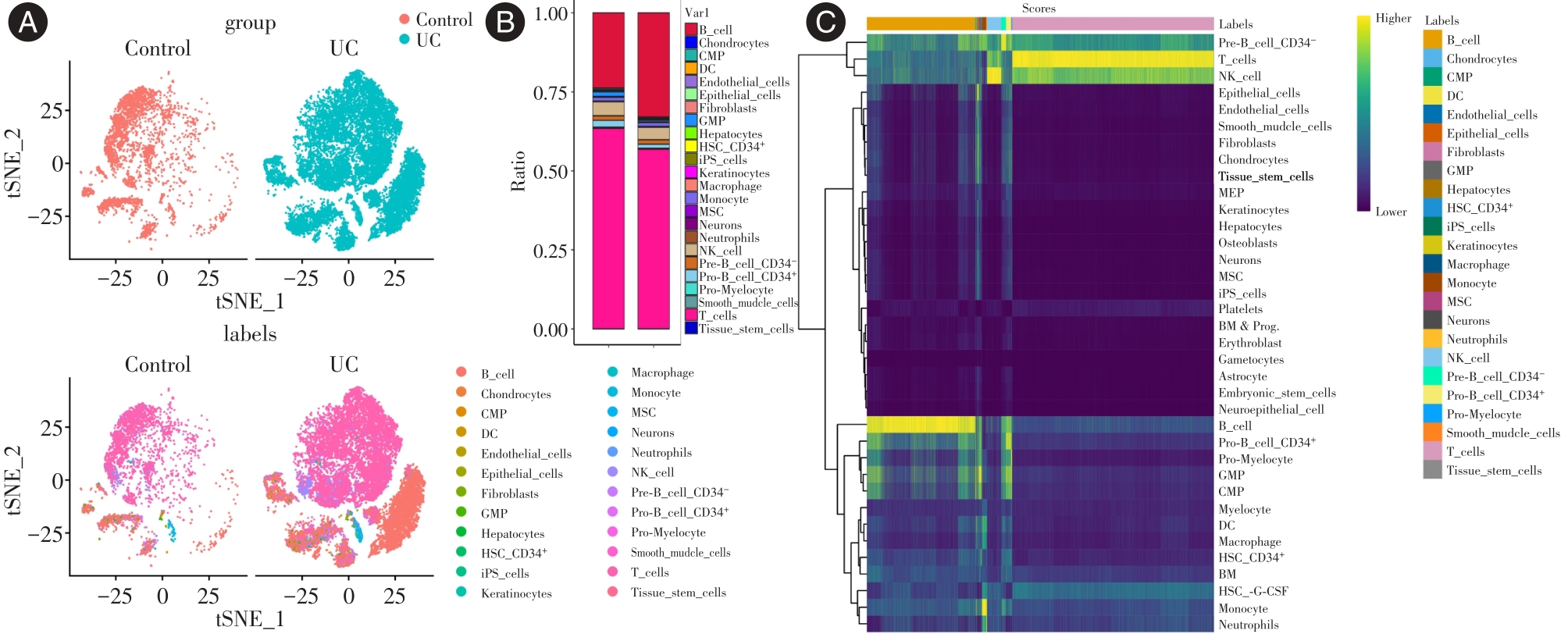
| 1 |
SEGAL J P, LEBLANC J F, HART A L. Ulcerative colitis: an update[J]. Clin Med (Lond), 2021,21(2):135-139. doi:10.7861/clinmed.2021-0080
doi: 10.7861/clinmed.2021-0080
|
| 2 |
PASVOL T J, HORSFALL L, BLOOM S, et al. Incidence and prevalence of inflammatory bowel disease in UK primary care: a population-based cohort study[J]. BMJ Open, 2020,10(7):e036584. doi:10.1136/bmjopen-2019-036584
doi: 10.1136/bmjopen-2019-036584
|
| 3 |
QIAO Y, RAN Z. Potential influential factors on incidence and prevalence of inflammatory bowel disease in mainland China[J]. JGH Open, 2019,4(1):11-15. doi:10.1002/jgh3.12238
doi: 10.1002/jgh3.12238
|
| 4 |
KOBAYASHI T, SIEGMUND B, LE BERRE C, et al. Ulcerative colitis[J]. Nat Rev Dis Primers, 2020,6(1):74. doi:10.1038/s41572-020-0205-x
doi: 10.1038/s41572-020-0205-x
|
| 5 |
YU Y R, RODRIGUEZ J R. Clinical presentation of Crohn's, ulcerative colitis, and indeterminate colitis: Symptoms, extraintestinal manifestations, and disease phenotypes[J]. Semin Pediatr Surg, 2017,26(6):349-355. doi:10.1053/j.sempedsurg.2017.10.003
doi: 10.1053/j.sempedsurg.2017.10.003
|
| 6 |
UNGARO R, COLOMBEL J F, LISSOOS T, et al. A Treat-to-Target Update in Ulcerative Colitis: A Systematic Review[J]. Am J Gastroenterol, 2019,114(6):874-883. doi:10.14309/ajg.0000000000000183
doi: 10.14309/ajg.0000000000000183
|
| 7 |
BOLAND B S, HE Z, TSAI M S, et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses[J]. Sci Immunol, 2020,5(50):eabb4432. doi:10.1126/sciimmunol.abb4432
doi: 10.1126/sciimmunol.abb4432
|
| 8 |
SATIJA R, FARRELL J A, GENNERT D, et al. Spatial reconstruction of single-cell gene expression data[J]. Nat Biotechnol, 2015,33(5):495-502. doi:10.1038/nbt.3192
doi: 10.1038/nbt.3192
|
| 9 |
LIBERZON A, BIRGER C, THORVALDSDÓTTIR H, et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection[J]. Cell Syst, 2015,1(6):417-425. doi:10.1016/j.cels.2015.12.004
doi: 10.1016/j.cels.2015.12.004
|
| 10 |
张士伟,程慎令,邢启峰. 茯苓酸通过抑制cGAS-STING信号通路减轻溃疡性结肠炎大鼠结肠上皮细胞损伤[J]. 免疫学杂志, 2023, 39(8): 672-680.
|
| 11 |
ALI A, TAN H, KAIKO G E. Role of the Intestinal Epithelium and Its Interaction With the Microbiota in Food Allergy[J]. Front Immunol, 2020,11:604054. doi:10.3389/fimmu.2020.604054
doi: 10.3389/fimmu.2020.604054
|
| 12 |
TSANG D K L, WANG R J, DE SA O, et al. A single cell survey of the microbial impacts on the mouse small intestinal epithelium[J]. Gut Microbes, 2022,14(1):2108281. doi:10.1080/19490976.2022.2108281
doi: 10.1080/19490976.2022.2108281
|
| 13 |
SAYOC-BECERRA A, KRISHNAN M, FAN S, et al. The JAK-Inhibitor Tofacitinib Rescues Human Intestinal Epithelial Cells and Colonoids from Cytokine-Induced Barrier Dysfunction[J]. Inflamm Bowel Dis, 2020,26(3):407-422. doi:10.1093/ibd/izz266
doi: 10.1093/ibd/izz266
|
| 14 |
EASTAFF-LEUNG N, MABARRACK N, BARBOUR A, et al. Foxp3+ regulatory T cells, Th17 effector cells, and cytokine environment in inflammatory bowel disease[J]. J Clin Immunol, 2010,30(1):80-89. doi:10.1007/s10875-009-9345-1
doi: 10.1007/s10875-009-9345-1
|
| 15 |
UZZAN M, MARTIN J C, MESIN L, et al. Ulcerative colitis is characterized by a plasmablast-skewed humoral response associated with disease activity[J]. Nat Med, 2022,28(4):766-779. doi:10.1038/s41591-022-01680-y
doi: 10.1038/s41591-022-01680-y
|
| 16 |
王小天,王月,彪雅宁,等. 燮理汤对溃疡性结肠炎小鼠TLR4/NF-κB/HIF-1α通路的影响[J]. 中国实验方剂学杂志,2023,29(8):142-149.
|
| 17 |
高飞,徐琪,林怡,等. 柴胡皂苷A对溃疡性结肠炎大鼠IKK/IKB/NF-κB信号通路及肠上皮细胞凋亡的影响[J]. 中国老年学杂志,2022,42(17):4289-4294. doi:10.3969/j.issn.1005-9202.2022.17.041
doi: 10.3969/j.issn.1005-9202.2022.17.041
|
| 18 |
DONG G, YANG Y, ZHANG H, et al. Protein Kinase CK2 Maintains Reciprocal Balance Between Th17 and Treg Cells in the Pathogenesis of UC[J]. Inflamm Bowel Dis, 2022,28(6):830-842. doi:10.1093/ibd/izab312
doi: 10.1093/ibd/izab312
|
| 19 |
WANG M, HUANG X, KANG Z, et al. Mechanism of Sishen-Pill-Regulated Special Memory T and mTfh Cell via Involving JAK/STAT5 Pathway in Colitis Mice[J]. Evid Based Complement Alternat Med, 2022,2022:6446674. doi:10.1155/2022/6446674
doi: 10.1155/2022/6446674
|
| 20 |
刘滨,刘雅清,宋红新,等. 黄芩汤对溃疡性结肠炎小鼠Th17/Treg、Th1/Th2细胞平衡的影响[J]. 中国实验方剂学杂志,2022,28(22):7-15.
|
| 21 |
LI X, LIU X, ZHANG Y, et al. Protective effect of Gloeostereum incarnatum on ulcerative colitis via modulation of Nrf2/NF-κB signaling in C57BL/6 mice[J]. Mol Med Rep, 2020,22(4):3418-3428.
|
| 22 |
WANG H, ZHOU Q, XIE D F, et al. LAPTM4B-mediated hepatocellular carcinoma stem cell proliferation and MDSC migration: implications for HCC progression and sensitivity to PD-L1 monoclonal antibody therapy[J]. Cell Death Dis,2024,15(2):165. doi:10.1038/s41419-024-06542-8
doi: 10.1038/s41419-024-06542-8
|
| 23 |
DING Y, JIANG Y, ZENG H, et al. Identification of a robust biomarker LAPTM4A for glioma based on comprehensive computational biology and experimental verification[J]. Aging (Albany NY), 2024,16. doi: 10.18632/aging.205736 .
doi: 10.18632/aging.205736
|
| 24 |
PAK Y, GLOWACKA W K, BRUCE M C, et al. Transport of LAPTM5 to lysosomes requires association with the ubiquitin ligase Nedd4, but not LAPTM5 ubiquitination[J]. J Cell Biol, 2006,175(4):631-645. doi:10.1083/jcb.200603001
doi: 10.1083/jcb.200603001
|
| 25 |
KAWANO Y, OUCHIDA R, WANG J Y, et al. A novel mechanism for the autonomous termination of pre-B cell receptor expression via induction of lysosome-associated protein transmembrane 5[J]. Mol Cell Biol, 2012,32(21):4462-4471. doi:10.1128/mcb.00531-12
doi: 10.1128/mcb.00531-12
|
| 26 |
ZOUALI M. Transcriptional and metabolic pre-B cell receptor-mediated checkpoints: implications for autoimmune diseases[J]. Mol Immunol, 2014,62(2):315-320. doi:10.1016/j.molimm.2014.01.009
doi: 10.1016/j.molimm.2014.01.009
|
| 27 |
SONG Z, WANG X, HE L, et al. Suppression of lysosomal-associated protein transmembrane 5 ameliorates cardiac function and inflammatory response by inhibiting the nuclear factor-kappa B (NF-κB) pathway after myocardial infarction in mice[J]. Exp Anim, 2022,71(4):415-425. doi:10.1538/expanim.22-0008
doi: 10.1538/expanim.22-0008
|